Technology
Appropriate models for heterogeneous multi-gene data sites
 2013-04-15
2013-04-15
Download
Right click and do "save link as"
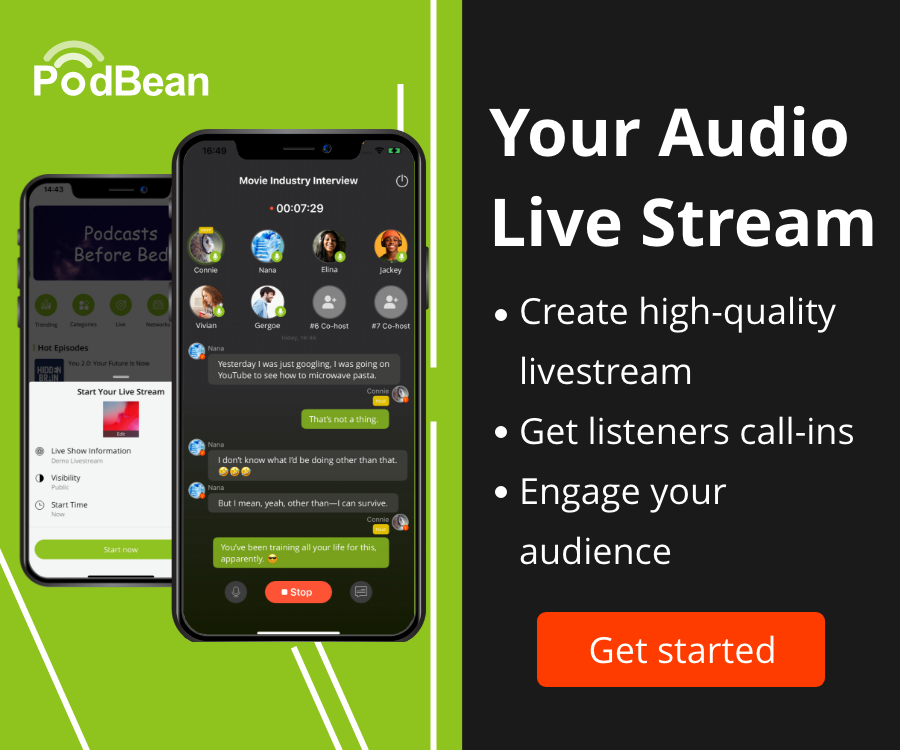
Multigene data sets are becoming the norm for phylogenetic studies; so called ‘phylogenomic datasets’ may involve hundreds of genes for many species. These data sets create challenges for current phylogenetic methods, as different genes have different functions and hence evolve under different processes. The question is how best to model this heterogeneity to give reliable phylogenetic estimates of the species tree.
Here we discuss two approaches. The first approach involves stochastic partitioning of genes into different classes, the second approach is a form of penalised maximum likelihood where the penalty term 'encourages' parameter estimates to be similar between different gene trees.
view more
More Episodes
10 Men and 1 woman?
 2016-06-01
2016-06-01
2016-06-01
Tripartitions in phylogenetic networks
2013-04-15
2013-04-15
Untangling tanglegrams
2013-04-15
2013-04-15
Some broad questions about the tree of life
2013-04-15
2013-04-15
Phylogenetic models and algebra
2013-04-15
2013-04-15
Phylogenetic networks
2013-04-15
2013-04-15

Get this podcast on your
phone, FREE